Maths/industry workshop on molecular simulation for pharmaceutical industry (Friday, April 28th)
A one-day workshop is organized on Friday April 28th (after the 3-day workshop on numerical methods for nonequilibrium systems), on the use of computational statistical physics in the pharmaceutical industry and related fields.
It will take place at IHP, lecturing room Darboux.
This event is organized by Chris Chipot (CNRS & Universite de Lorraine), Tony Lelièvre, Gabriel Stoltz (Ecole des Ponts & Inria Paris) and benefits from the framework of the LIA between the Centre National de la Recherche Scientifique and the University of Illinois at Urbana-Champaign. It is also funded by AMIES (french agency to foster interactions between maths and industry) and the ERC MsMaths.
The workshop consists of 5 lectures by researchers from the chemical and pharmaceutical industries (BASF, Boehringer, CEA, GSK, Johnson & Johnson, Sanofi), as well as one short lecture illustrating the point-of-view of mathematicians. It will start at 9h30 and end around 17h00, with lunch buffet.
You can register here.
Program (abstracts below)
- 09h15 - 09h30 Welcome
- 09h30 - 10h30 Thomas Fox (Boehringer-Ingelheim) [slides]
Molecular Dynamics Simulations in Drug Discovery - 10h30 - 11h00 Coffee break
- 11h00 - 12h00 Henrik Keränen (Johnson & Johnson)
Free energy calculations in drug discovery - 12h00 - 13h00 Marc Bianciotto (Sanofi)
Shooting a moving target: simulation of ligand binding to flexible proteins - 13h00 - 14h15 Lunch buffet
- 14h15 - 15h15 Pieter in‘t Veld (BASF)
Soft matter modeling in an industrial environment - 15h15 - 16h15 Michel Masella (CEA) [slides]
An O(N) multi-scale N-body approach for simulating polarizable microscopic systems - 16h15 - 16h45 Yvon Maday (UPMC and Carnot Smiles)
Collaboration between experts in maths and computational chemistry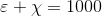
- 16h45 Closing coffee time

Thomas Fox, Molecular Dynamics Simulations in Drug Discovery
I will give a short overview over the drug discovery process at a typical pharmaceutical company, then I will discuss two applications of MD simulations to support this process: With mixed-solvent MD simulations we map out favorable interactions on the surface of proteins to support the finding and optimization of small ligands binding to the protein. A second application is the determination of relative free energies of binding of small ligands to a protein with Thermodynamic Integration - here I discuss recent advances and point to potential problems of this approach.
Michel Masella, An O(N) multi-scale N-body approach for simulating polarizable microscopic systems
The aim of our group is to propose alternative routes to tackle the limitations that are inferred to be at the origin of the relatively modest role of MD simulations to develop new drugs. In particular, we are developing (1) a new generation of force fields explicitly accounting for polarization and “charge transfer” effects; (2) an O(N) multi-scale coarse grained approach to efficiently handle large chemical environments; and (3) a parallel simulation code POLARIS(MD), where the latter theoretical approaches (as well as a scalable O(N) Fast Multipole Method) are implemented. We will present here these different approaches and one of their applications to simulate a million atom scale molecular system (namely a virus capsid solvated in liquid water) using overall modest computational ressources.